Auftragsarbeit
Soweit unser Lehr- und Forschungsauftrag dies erlaubt, sind wir neben der Kooperation mit anderen wissenschaftlichen Einrichtungen auch gerne bereit, Unternehmen der Privatwirtschaft mit unserem Knowhow und unseren Messeinrichtungen bei bodenkundlichen Fragestellungen zu unterstützen.
Laborausstattung
Strukturelle Verfahren
-
FT-IR: Fourier Transformations Infrarot Spektroskop
![]()
![]()
![]()
FTIR-Spektrometer (Tensor 27, Bruker)
Die IR-Spektroskopie wird typischerweise zur Untersuchung der Struktur von organischen Bestandteilen und Feststoffen wie Mineralen eingesetzt. Bei Vorliegen bekannter Substanzen kann das Verfahren auch für die Quantifizierung eingesetzt werden. Da die Verhältnisse auf molekularer Ebene erfasst werden, können z.B die Veränderung struktureller Verhältnisse durch die Sorption einer organischen Substanz an einer Festphase untersucht werden. IR vermittelt Angaben über den Nahbereich von Strukturen die röntgenographisch nicht zu erfassen sind.
Atome werden im molekularen Bereich durch elastische Bindungen zusammengehalten. Der elastische Charakter der Bindungen ermöglicht Schwingungen der Teilchen um eine Ruhelage. Liegen die Schwingungen im Bereich der Wellenzahl 4000-400 cm-1, sind sie der Analyse mit dem Infrarot (IR) zugänglich. Die Frequenz der Schwingungen hängt von der atomaren Masse, den Bindungskräften und der räumlichen Anordnung der Teilchen ab. Als Strahlungsquelle für den mittleren und fernen Infrarot-Bereich dient ein Globar, ein aus Siliziumcarbid bestehender Stab der elektrisch erhitzt wird. Es werden Absorptionsbanden erhalten, die typisch für die jeweiligen Stoffe/Stoffgruppen sind.
Mit dem IR können orientierte oder texturfreie Präparate im Transmissions- oder Reflexionsmodus gemessen werden. Die Korngröße einer zu untersuchenden Substanz sollte kleiner als 2 µm sein, um den Christiansen-Effekt zu vermeiden. Die Nachweisgrenze der Methode ist niedrig. Bei Linearität zwischen Extinktion und Konzentration ist Quantifizierung möglich. Eine Klimakammer ermöglicht in situ Messungen bei verschiedenen Drücken und Temperaturen im DRIFT-Modus für streuendes Probenmaterial (DRIFT: diffuse reflectance infrared fourier transform). Es kann auch eine Zelle an die ATR-Einheit ( ATR: attenuated total reflection, deutsch: abgeschwächte Totalreflexion) adaptiert werden, um den Transport in porösen Materialen mit Austauschexperimenten zu untersuchen.
-
Goniometermikroskop
![Bild des Gerätes]()
![Bild des Gerätes]()
![Bild des Gerätes]()
![Bild zweier Tropfen]()
![Bild zweier Tropfen]()
![Bild zweier Tropfen]()
Kontaktwinkelmikroskop (OCA 20, DataPhysics, Filderstadt)
Die Benetzungseigenschaften eines Bodens, also das Verhalten gegenüber Wasser, wirken sich auf die hydraulischen Funktionen (z.B Infiltration, Wasserverteilung) und damit korrelierte Parameter wie Pflanzenwachstum oder mikrobiellen Abbau aus. Man unterscheidet spontanes Spreiten (CA=0°; „hydrophil“), nicht spontanes Spreiten (CA>0<90°; „subkritische Benetzungshemmung“) und Tropfenbildung (CA≥90°). Bestimmt werden die Benetzungseigenschaften häufig über den Kontaktwinkel (CA), der sich an der 3-Phasen-Grenze fest/flüssig/gasförmig ausbildet.
Mit dem Kontaktwinkelmikroskop werden CA nach der sessile drop-Methode (SDM) direkt durch Anlegen von Tangenten auf beiden Seiten eines auf die Probenoberfläche aufgesetzten Tropfens bestimmt. Die Verwendung von Flüssigkeiten mit verschiedenen Oberflächenspannungen und Polaritäten erlaubt die Berechnung der freien Oberflächenenergie (SFE).Mit einer Videokamera werden das Aufsetzen eines Tropfens auf die Probenoberfläche und die zeitliche Veränderung des Kontaktwinkels aufgezeichnet. Der CA wird mit der Software SCA20 (DataPhysics, Filderstadt) direkt nach dem Aufsetzen („initialer CA“) und nach (üblicherweise) 1 Sekunde und 5 Sekunden bestimmt. Die Probe ist mit doppelseitigen Klebeband auf einem Objektträger fixiert (idealerweise eine ein-Korn-Schicht zur Vermeidung von Kapillarkräften).
Für die SDM eignen sich pulverförmige trockene Proben, die mit doppel-seitigem Klebeband fixiert sind, aber auch andere nicht-poröse Oberflächen (z.B. Glas mit definiert veränderten Oberflächeneigenschaften). Tropfen können auch auf einzelne Aggregate aufgesetzt werden. Durch Verwendung sehr dünner Dosiernadeln ist der CA einzelner Mineralkörner bestimmbar und so eine hohe räumliche Auflösung der Benetzungseigenschaften erreichbar. Auch eine Transekt-weise Beprobung von Bodenprofilen (trocken!) unter Konservierung der Anordnung der Körner ist durch direktes Andrücken von Klebeband möglich. Die SDM deckt den gesamten CA-Bereich (theoretisch 0-180°) ab.
-
XPS: Photoelektronenspektroskop mit Auger-Einheit
![Bild des Gerätes]()
![Bild des Gerätes]()
![Bild des Gerätes]()
Kratos Axis Ultra DLD
Die Röntgenphotoelektronenspektro-skopie (XPS) erlaubt die Analyse der chemischen Zusammensetzung (außer H und He) von organischen und anorganischen Oberflächen sowie die Analyse von chemischen Bindungszu-ständen. Die Informationstiefe liegt dabei bei rd. 1−10 nm. Die XPS-Analyse von Oberflächen kann als Punktanalyse (~10 µm), als 2-D Image (laterale Pixelauflösung ~3 µm) oder entlang von definierten Linienprofilen erfolgen. Die Kombination mit Sputtertechniken (mittels Argon oder Coronen) erlaubt zudem tiefenaufgelöste Elementanalysen. Die XPS-Analytik dient vorrangig der Charakterisierung von organischen Belägen auf Mineraloberflächen (C und N Spezies), der Quantifizierung von Mineralverwitter-ungsprozessen, sowie der Untersuchung von Redox-transformationen mineralischer und organischer Phasen.
Die Messung der Elementzusammensetzung und Bindungszustände beruht auf dem Photoeffekt, bei dem mittels Anregung durch Photonen (Röntgenstrahlung, AlKa oder MgKa) Rumpfelektronen aus Atomen, Molekülen oder Festkörpern herausgelöst werden. Die kinetische Energie der erzeugten Photoelektronen (Ekin) steht dabei in direktem Zusammenhang zur elementspezifischen Bindungsenergie (Ebinding) der Elektronen gemäß Ebinding = Ephoton * (Ekin + Ø), wobei Ephoton der Energie der Röntgenphotonen und Ø der Arbeitsfunktion des Spektrometers entspricht. Die chemische Verschiebung in den Bindungsenergien der Elektronen eines Elementes gibt Auskunft über deren Bindungsumgebung, d.h. über den Oxidationszustand eines Elementes bzw. die Gegenwart bestimmter funktioneller Gruppen.
Eine Aufladung des Probenmaterials wird durch Einspeisung niedrig energetischer Elektronen verhindert, sodass neben leitenden auch nichtleitende Materialien wie etwa Gläser, Minerale, Gesteine oder biologische Materialien untersucht werden können. Eine Cryo-Einheit, in der die Probe in wenigen Sekunden bei ca. -130°C gefroren wird, erlaubt ferner die in-situ Analyse von trocknungsanfälligen Materialien. Zu untersuchende Proben müssen im Ultrahochvakuum stabil sein.




Quantitative Verfahren
-
BET-Oberflächenanalysator
![Bild des Gerätes]()
![Bild des Gerätes]()
![Bild des Gerätes]()
Autosorb-1 und Nova4000e
Mithilfe unserer Oberflächenanalysatoren lassen sich Aussagen über die Größe von Festphasenoberflächen und Porenverteilung auf Nanometer-Ebene treffen. Standardgrößen zur Charak-terisierung der Bodenfestphasen beinhalten neben der BET Oberfläche das Gesamtporenvolumen (in Poren <rd. 200 nm) sowie die Volumina bzw. Oberflächen in Mikro- (<2 nm), Meso- (2–50 nm) und Makroporen (>50 nm). Mittels des Autosorb-1 lässt sich außerdem die Porenverteilung von sehr mikroporösen Materialien, z.B. Zeolithe, Tonminerale, Metalloxide sowie Kohlen u.ä., bei sehr niedrigen Relativdrücken untersuchen. Die Kenndaten liefern dabei wichtige Hinweise auf die Reaktivität der Bodenfestphasen, z.B., über deren Fähigkeit, organische Substanz bzw. anorganische Bodenlösungsbestandteile (z.B. Metalle) zu binden.
Die Messung der spezifischen Oberflächen und Porosität erfolgt durch Physisorption gasförmigen Stickstoffs bei 77 K als Funktion des Druckes. Durch die Messung bei verschiedenen Gleichgewichtsdampfdrücken können Adsorptions- als auch Desorptionsisothermen in einem relativen Druckbereich ab 5 × 10–3 bis ca. 1.0 bar aufgenommen werden. Hierfür wird ein definiertes Gasvolumen zur Probe dosiert und nach Gleichgewichtseinstellung der Gasdruck in der Messzelle gemessen (volumetrisch-statische Messmethode). Die Adsorptions- und Desorptionsisothermen bilden die Grundlage zur Ermittlung der spezifischen Oberfläche (z.B. BET Methode) bzw. der Porenverteilungen (z.B. gemäß V-t, BJH, DR).
Die zu untersuchenden Proben müssen wasserfrei sein, was durch einen vorgeschalteten Trocknungsprozess im Vakuum bei 40–200°C gewährleistet wird. Für die Gasadsorptionsmessung selbst werden mindestens 5–10 m2 Oberfläche je Messzelle benötigt. Bei groben, makroporösen Materialien, z.B. Gesteinsfragmenten, gestaltet sich daher eine zuverlässige Messung schwierig.
-
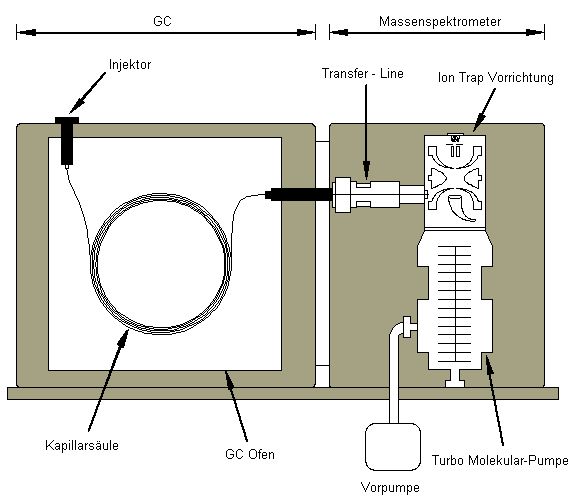
GC-MS: Massenspektrometer (Iontrap) in Kopplung mit einem Gaschromatographen
![Bild des Gerätes]()
![Bild des Gerätes]()
![Bild des Gerätes]()
![Schematischer Aufbau]()
![Schematischer Aufbau]()
![Schematischer Aufbau]()
GC-MS (Varian 450GC-220MS - Iontrap)
Über die Kopplung des Massenspektrometers mit einem Gaschromatographen können org. Moleküle (Monomere), die in org. Extraktionsmitteln gelöst sind, voneinander separiert und einzeln identifiziert und quantifiziert werden.
Org. Monomere werden in der Bodenkunde vielfach als Repräsentanten von Umsatzprozesse oder org. Stoffgruppen / Bodenpools einer höheren hierarchischen Ebene verwendet (Biomarker). Die Verwendung von Biomarkern ermöglicht es, die Dynamik definierter org. Bodenpools qualitativ und quantitativ zu untersuchen und deren Zusammensetzung halbquantitativ zu erfassen.
Da es z.B. kaum möglich ist, pflanzliche Kohlenstoffeinträge von mikrobiellem Kohlenstoff (C) in seiner Gesamtheit differenziert zu quantifizieren, werden pflanzliche und mikrobielle Zucker als Repräsentanten dieser C-Pools verwendet. Die mikrobielle Zersetzergemeinschaft kann hinsichtlich ihrer Zusammensetzung aus ökophysiologisch unterschiedlichen Gruppen mittels Phospholipidfettsäure-Analyse untersucht werdenFunktionsweise
1) Die einzelnen org. Moleküle eines Probenextraktes werden mittels GC (Abb.2) voneinander getrennt. Dazu wird die Probe in einem Injektor bei ca. 250 - 300°C verdampft und das entstehende Gas mittels Trägergas (Helium) in eine angeschlossene chromatographische Trennsäule gespült. Je nach chemischen Eigenschaften und Molekülgröße werden die org. Analyten auf der inneren Beschichtung dieser Trennsäule unterschiedlich stark sorbiert. Durch anschließende Erhöhung der Säulentemperatur werden die Analyten zu unterschiedlichen Zeitpunkten desorbiert und verlassen damit zu unterschiedlichen Zeitpunkten die Säule in Richtung Detektor.
2) Im Detektor werden die Einzelsubstanzen durch Kollision mit Elektronen (el. Ionisierung) oder mit Molekülen eines Ionisierungsgases ionisiert (chem. Ionisierung). Dabei zerfallen die Moleküle in positiv geladen Bruchstücke, die in einem el. Feld (Iontrap) stabil gehalten werden. Durch Erhöhung der Spannung werden dann einzelne Bruchstücke je nach ihrer Ladung und Ionenmasse destabilisiert und Richtung Detektor abgelenkt, wo sie beim Einschlag eine Elektronenkaskade auslösen, welche als messbarer Strom das Messsignal darstellt. Dessen Intensität ermöglicht die Quantifizierung des Ausgangsmoleküls, während die Bruchstückmuster der bei der Ionisierung entstehenden Ionen Aussagen über die Struktur des Moleküls zulassen (Substanzidentifikation).Im Prinzip können mit dem GC-MS alle org. Moleküle separiert und detektiert werden.
Eine erste grundsätzliche Limitation besteht allerdings bei der Volatilisation von Substanzen im Injektor. Ist diese nicht möglich, weil die Substanzen oder ihre Derivate nicht flüchtig genug sind, ist eine Analyse nicht möglich. Bei der Detektion stellt v.a. die Molekülgröße eine Limitation dar, da die größte detektierbare Masse 650 u ist.
-
HYPROP
Mit dem HYPROP-System können die Retentionskurve und die Leitfähigkeit bei Teilsättigung berechnet werden. Die ungestörten Proben in Stechzylindern werden hierzu auf Messköpfe mit zwei unterschiedlich langen Tensiometern aufgesetzt und durch Verdunstung sukzessive entwässert, eventuell wird die Verdunstung durch Ventilatoren verstärkt um Zeit zu sparen. Die Daten der Tensiometer werden in Zeitintervallen von einem Computerprogramm aufgenommen und abgespeichert, das Gewicht wird halbmanuell abgefragt und ebenfalls gespeichert. Nach Abschluss der Entwässerung werden die Daten in die HYPROP-Auswerte-Software übertragen, die dann die van Genuchten-Parameter sowie die Parameter für die Wasserleitfähigkeit bei Teilsättigung liefert.
-
IC: Ionenchromatograph
![Bild des Gerätes]()
![Bild des Gerätes]()
![Bild des Gerätes]()
ICS 90 Dionex/ Thermo Fisher Scientific
Es können hiermit in einem Analysengang die Konzentra-tionen an Anionen und Kationen in wässrigen Lösungen gemessen werden. Dabei kann es sich z.B. um Bodenextrakte für die Untersuchung des Nährstoffgehaltes von Bodenproben handeln oder um Grundwasserproben, um die Verlagerung von Ionen mit dem Sickerwasser in tiefere Bodenschichten zu bestimmen.
Bei der Ionenchromatographie werden die in einer wässrigen Lösung enthaltenen Ionen („mobile“ Phase) in Kontakt mit einer „stationären“ Phase, die sich in einer Säule befindet, gebracht. Die Auftrennung erfolgt, wenn die Einzelsubstanzen beim Transport über bzw. durch die stationäre Phase unterschiedlich stark mit dieser in Wechselwirkung treten und somit unterschiedlich schnell wandern. Antreibende Kraft ist ein Druckgradient, der über einen sogenannten Eluenten aufgebaut wird. Die Säule ist mit Ionentauschern gefüllt, entweder Anionentauscher, die Anionen aus der Probe gegen OH-- oder HCO3--Ionen tauschen (Anionenchromatographie), oder Kationentauscher, die Kationen aus der Probe gegen H+-Ionen tauschen können (Kationenchromatographie). Diese Bindung ist grundsätzlich reversibel, und ein Rücktausch erfolgt, wenn aus dem entsprechend angesetzten Eluenten OH-- bzw. HCO3--Ionen respektive Protonen nachgeliefert werden. Allerdings ist für die einzelnen Komponenten der Probenlösung die Festigkeit der Bindung und damit die Geschwindigkeit des Rücktauschs aufgrund ihrer Oberflächenladungsdichten und anderer physikochemischen Eigenschaften stark unterschiedlich. Hierdurch werden die verschiedenen Ionenspezies im Laufe einer Analyse voneinander getrennt. Eine vollständige Trennung ist wichtig, um die zu messenden Ionen einzeln quantifizieren zu können: Hier erfolgt die Quantifizierung über eine elektrische Leitfähigkeitsmesszelle, durch die die Probe fließt. Das Leitfähigkeitssignal wird kontinuierlich aufgenommen und gegen die Zeit aufgetragen. Es ergibt sich eine Kurve („Peak“), die computergestützt integriert wird. Die Peakfläche ist im typischen Messbereich von ca. 0.1-0.5 (je nach Ion) bis 50 mg L-1 proportional zur Stoffkonzentration (Kalibration mit etwa 5 Standards bekannter Konzentrationen). Die Zuordnung der einzelnen, nacheinander aufgenommenen Messsignale zu den verschiedenen Ionen erfolgt über die Retentionszeit, welche von den Messungen der Standards her bekannt ist.
Gemessen werden können u.a. Nitrat, Phosphat, Sulfat, Chlorid, Bromid, Jodid, Acetat, Oxalat, Ammonium, Kalium, Natrium, Calcium, Magnesium. Die Proben müssen vorweg feinfiltriert werden (< 0.45 µm). Problematisch ist die Messung dann, wenn die Konzentrationen der einzelnen Ionen sehr unterschiedlich sind und die Peaks der Ionen mit hoher Konzentration andere Peaks überlagern, z.B. bei hohen Chlorid- oder Sulfatgehalten oder bei mit HNO3 oder HCl angesäuerten Proben.
-
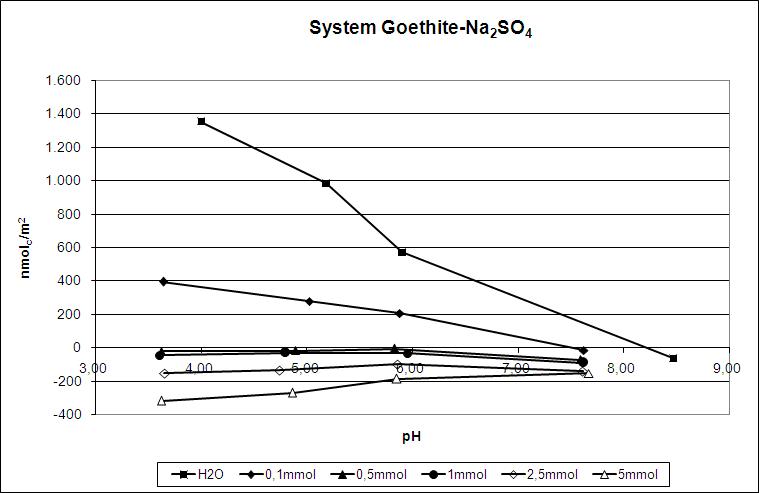
Particle-Charge-Analyzer
![Bild des Gerätes]()
![Bild des Gerätes]()
![Bild des Gerätes]()
![Grafik]()
![Grafik]()
![Grafik]()
Partikelladungsdetektor (PCD 03, Fa. Mütek)
Die Oberflächenladung an der fest/flüssig-Grenzfläche ist im Zusammenhang mit dem Sorptions-vermögen von Stoffen für geladene und ungeladene Teilchen, der Stabilität der Aggregierung von z.B. Tonfraktionen aus Böden und der Löslichkeit von Mineralen eine gesuchte Größe. Mit dem Partikelladungsdetektor kann die Oberflächenladung von feinkörnigen Teilchen wie Tonmineralen, gelöster organischer Substanz und Oxiden des Fe, Al und Mn bei verschiedenen pH-Werten und Lösungszusammensetzungen analysiert werden. Im Unterschied zu der Bestimmung des Zetapotentials erlaubt die hier eingesetzte Untersuchung des Strömungspotentials kombiniert mit Polyelektrolyttitration bis zum Ladungsnullpunkt die Quantifizierung der an den äußeren Oberflächen vorhandenen Ladungen.
Die Oberflächenladung von z.B. der Tonfraktion von Böden, oder allgemein Teilchen in Suspension, wird im Partikelladungsdetektor über einen elektrokinetischen Effekt bestimmt. Ein mit 3 Hz oszillierender PTFE-Kolben versetzt die Probensuspension in Bewegung und verursacht dadurch ein Strömungspotential, das über zwei in die PTFE-Meßzelle eingebaute Au-Elektroden abgegriffen wird. Negative Oberflächenladung wird durch Titration mit einer Standardlösung des kationischen Polyelektrolyten poly-Diallyl-dimethyl-ammoniumchlorid ,,Poly-DADMAC quantitativ bestimmt. Die Ladungstitration der Suspensionen erfolgt durch Zugabe der Titrationslösung über den isoelektrischen Punkt hinaus. Der Titrationsendpunkt wird bei Potential Null festgelegt. Positive Oberflächenladung wird mit Hilfe eines anionischen Polyelektrolyten quantifiziert. Während der Titration werden das Strömungspotential, der pH-Wert und die zutitrierte Menge fortlaufend aufgezeichnet.
In die Messzelle können Suspensionen mit Teilchendurchmessern bis ca. 6 µm eingebracht werden. Der Materialbedarf ist mit ca. 10 mg pro Messung relativ gering. Neben den Verhältnissen an der Oberfläche von festen Partikeln wie Tonmineralen und pedogenen Oxiden sind auch die Ladungseigenschaften von verschiedenen gelösten organischen Substanzen durch das Strömungspotential zu erhalten und durch Polyelektrolyttitration zu quantifizieren. Das Molgewicht der gelösten organischen Substanz darf für die Gewährleistung der Detektierbarkeit nicht zu gering sein.
-
Präzisionstensiometer
![Bild des Gerätes]()
![Bild des Gerätes]()
![Bild des Gerätes]()
Dynamisches Kontaktwinkeltensiometer (DCAT 11, DataPhysics, Filderstadt)
Die Benetzungseigenschaften eines Bodens, also das Verhalten gegenüber Wasser, wirken sich auf die hydrau-lischen Funktionen (z.B Infiltration, Wasserverteilung) und damit korrelierte Parameter wie Pflanzenwachstum oder mikrobiellen Abbau aus. Man unterscheidet spontanes Spreiten (CA=0°; „hydrophil“), nicht-spontanes Spreiten (CA>0<90°; „subkritische Benetzungshemmung“) und Tropfenbildung (CA≥90°). Bestimmt werden die Benetzungseigenschaften häufig über den Kontaktwinkel (CA), der sich an der 3-Phasen-Grenze fest/flüssig/gasförmig ausbildet.
Mit dem DCAT werden CA nach der Kapillaraufstiegsmethode (CRM) und der Wilhelmy-Platte-Methode (WPM) bestimmt. Die Verwendung von Flüssigkeiten mit verschiedenen Oberflächenspannungen und Polaritäten erlaubt die Berechnung der freien Oberflächenenergie (SFE). Außerdem kann mit dem DCAT die Oberflächenspannung von Flüssigkeiten mit der Wilhelmy-Platte oder dem DuNoüy-Ring gemessen werden.Das DCAT ist eine Präzisionswaage (Auflösung 10-5g), die die Gewichtszunahme der Probe als Funktion der Zeit aufzeichnet, bei der CRM durch kapillaren Aufstieg von H2O und n-Hexan (vollständig benetzende Flüssigkeit) in der Probe, bei der WPM durch Eintauchen eines Objektträgers, auf dem die Probe mit doppelseitigem Klebeband fixiert ist (Abb. 2). Der CA wird rechnerisch über die Steigung des linearen Bereichs der m2[g2]=f(t)-Kurve von H2O und n-Hexan (bzw. weiterer Flüssigkeiten) und die Flüssigkeitskonstanten (CRM), bzw. durch Extrapolation der Messkurve auf die Eintauchtiefe Null und Bestimmung der Benetzungskraft (WPM) ermittelt. Bei hydrophobem Material (CA>90°) kann die Wasseraufnahme über kapillaren Aufstieg als Funktion der Zeit als Langzeitmessung (>12h) bestimmt werden.
Für die CRM eignet sich sowohl pulverisiertes als auch aggregiertes Material. Auch können die Benetzungseigenschaften von Einzelaggregaten bestimmt werden. Naturgemäß ist die CRM auf CA<90° limitiert. Die Bestimmung der Flüssigkeitsaufnahme als Funktion der Zeit ist auch für massive poröse Probenkörper möglich (bis ca. 15g). Für CA-Bestimmungen von Bodenproben, bzw. körnigem Material allgemein mit der WPM müssen die Proben pulverförmig und trocken sein. Abgedeckt ist hier der gesamte CA-Bereich (theoretisch 0-180°). Möglich ist auch die CA-Bestimmung von definierten geometrischen Körpern mit nicht-poröser Oberfläche (z.B. Glasplatte mit definiert veränderten Oberflächeneigenschaften).
-
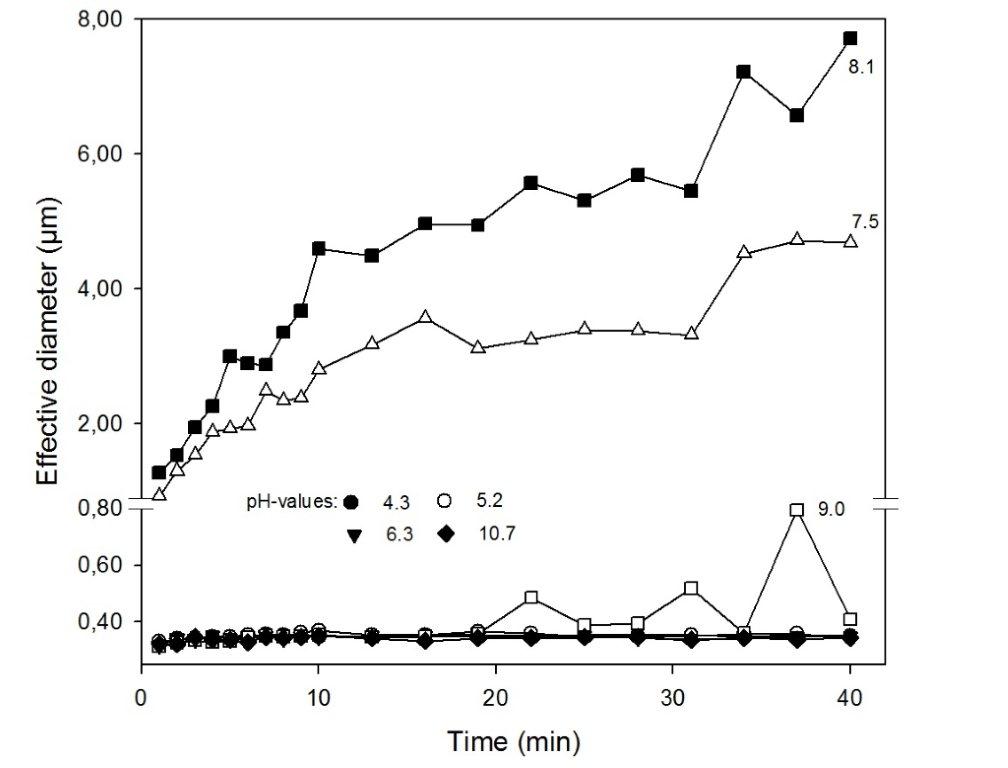
Zeta-Potential and Particle Size Analyzer
![Bild des Gerätes]()
![Bild des Gerätes]()
![Bild des Gerätes]()
![Grafik]()
![Grafik]()
![Grafik]()
Zeta-Potential- und Partikelgrößenmessgerät (Brookhaven, ZetaPals)
Das Zeta-Potential ist das elektrische Potential an der Abscherschicht eines in einer Suspension bewegten Partikels. Das auf Ladung beruhende elektrische Feld übt Kraft auf andere geladene Teilchen aus, die attraktiv bei unterschiedlichen Ladungsvorzeichen und repulsiv bei gleichnamigen Ladungen sind. Damit ist das Zeta-Potential eine wichtige Größe für die Wechselwirkung von Kolloiden z.B. von Tonmineralen im Porenwasser von Böden. Eisenoxide und organische Substanzen sind Stoffgruppen im Boden, die beide variable Ladungen aufweisen. Aufgrund des amphoteren Charakters der Eisenoxide weisen diese bei pH-Werten <6 eine positive Gesamtladung auf und interagieren dann mit negativ geladenen Partikeln wie (gelöster) organischer Substanz und Tonmineralen mit permanent negativer Ladung. Diese Bindung hat für die Stabilisierung organischer Substanz und durch die Vergrößerung der Partikel durch Aggregierung auch für deren Verlagerbarkeit im Sickerwasser erhebliche Bedeutung. Probenabhängig können mit dem Gerät im Größenbereich von <1 nm bis 6 µm die mittlere Teilchengröße und Standardabweichung volumengewichtet bestimmt werden.
Das Zeta-Potential wird mit Hilfe der ZetaPals-Technik über Phasen-Analyse-Lichtstreuung ermittelt, die weit empfindlicher ist als Methoden der Lichtstreuung, die auf einem verschobenem Frequenzspektrum basieren. Für die Bestimmung der Partikelgröße (hydrodynamischen Durchmessers) wird Streulicht genutzt, das entsteht, wenn Licht als elektromagnetische Welle beim Auftreffen auf einen Partikel Oszillationen erzeugt. Hierbei treten Fluktuationen der Streuintensität auf, die Basis für die „Quasi Elastische Licht Streuung (QELS)“ ist, die dem hier eingesetzten Messprinzip zugrunde liegt.
In die Messung werden bei Nutzung der Küvette (ohne Titrationseinheit) jeweils ca. 2 ml Suspension eingesetzt, die vorher auf bestimmte pH-Werte und Ionenkonzentrationen eingestellt werden kann. Während die Messung des Zeta-Potentials nur wenig von dem eingesetzten Feststoffgehalt der Suspension beeinflusst wird, ist für die Bestimmung der Partikelgröße die suspendierte Menge häufig von Relevanz. Auch muss bei der Bestimmung der Partikelgröße beachtet werden, dass sich der hydro-dynamische Durchmesser mit der Zeit verändern kann (siehe Abbildung).
Ein Titrator mit vier verschiedenen Kanälen kann für die automatische Bestimmung des isoelektrischen Punktes von verschiedenen kolloidalen Stoffgruppen eingesetzt werden. Auch kann die Titrationseinheit zur Untersuchung des Effektes der Salzkonzentration auf das Zeta-Potential genutzt werden. Bei der Nutzung des Titrators vergrößert sich aber die benötigte Suspensionsmenge gegenüber der Direktmessung in der Küvette.






Analytische Methoden
-
Texturanalyse
Körnung
Aus den Anteilen von Sand, Schluff und Ton einer Bodenprobe kann die Bodenart abgeschätzt werden, die einen Schlüssel Parameter der Bodenkunde darstellt. Die Korngrößenverteilung wird anhand von zwei kombinierten Methoden ermittelt. Die Sandfraktionen (Grob-, Mittel-, und Feinsand) werden anhand von Nasssiebung getrennt. Die Schlufffraktionen (Grob-, Mittel-, und Feinschluff) und die Tonfraktion werden anhand ihrer Sinkgeschwindigkeit in einem Fluid mit Hilfe einer speziellen Pipette getrennt. Die entsprechenden Massen werden nach der Trocknung bei 105°C bestimmt.
Diffusion-Tool
Referenz: Schwen and Böttcher 2013. A simple tool for the inverse estimation of soil gas diffusion coefficients. Soil Science Society of America Journal, doi:10.2136/sssaj2012.0347
Laborleitung


30419 Hannover





